Introduction
3DPatch is an application facilitating residue-level information content calculations, homology searches, and 3D structure conservation level-based mark-up for protein sequence or profile hidden Markov model (HMM) queries.
This section aims to provide an overview of how 3DPatch operates. Following sections provide more detail on individual steps in the data processing workflow and various website components.
3DPatch can accept either a single protein sequence or a profile HMM file as a new search input. Alternatively, results of previous 3DPatch sessions can be loaded from a special save point file. Given that the profile HMM file input route simply skips the first few steps of the sequence input-processing pipeline, this section illustrates most of the server functionality on an example utilizing the sequence input route. Loading the results of previous 3DPatch sessions is covered in a separate section.
After entering a valid protein sequence and clicking the submit button, the sequence is processed by the HMMER web server. There, a phmmer search against the UniProt reference proteomes sequence library is performed. In order to do this, the sequence input is initially converted to a profile HMM featuring position-independent parameters. HMMER web server performs a search using this profile HMM to find regions of sequences from the UniProt reference proteomes similar to the original sequence input.
Multiple sequence alignment (MSA) is obtained from the set of alignments of significant UniProt reference proteomes sequence hits to this primitive profile HMM. Second, higher-quality profile HMM is then built from this MSA, with parameters reflecting the frequencies of amino acid residues observed at individual positions in the MSA.
The profile HMM with position-specific parameters is used as an input to the hmmsearch program, which is used to find regions of sequences corresponding to 3D structures from the Protein Data Bank (PDB) similar to the original sequence input. At the same time, information content at individual positions in the profile HMM is calculated using the Skylign server. If a profile HMM file is used as an input to 3DPatch, previous steps are skipped and the submitted profile HMM is used directly as an input to hmmsearch and Skylign.
Mapping between individual residues in the identified PDB structures and positions in the profile HMM is obtained from the alignment of sequences corresponding to the respective 3D structures to the profile HMM. These alignments can be visualized in the sequence alignment window. Coverage of profile HMM positions by the PDB structures is illustrated using a few informative structures, and a list of other PDB structures containing a hit to the profile HMM is generated. Any of these structures can then be visualized with residue-level information content-based mark-up using the LiteMol viewer.
The input-processing workflow described above can be summarized in the following scheme: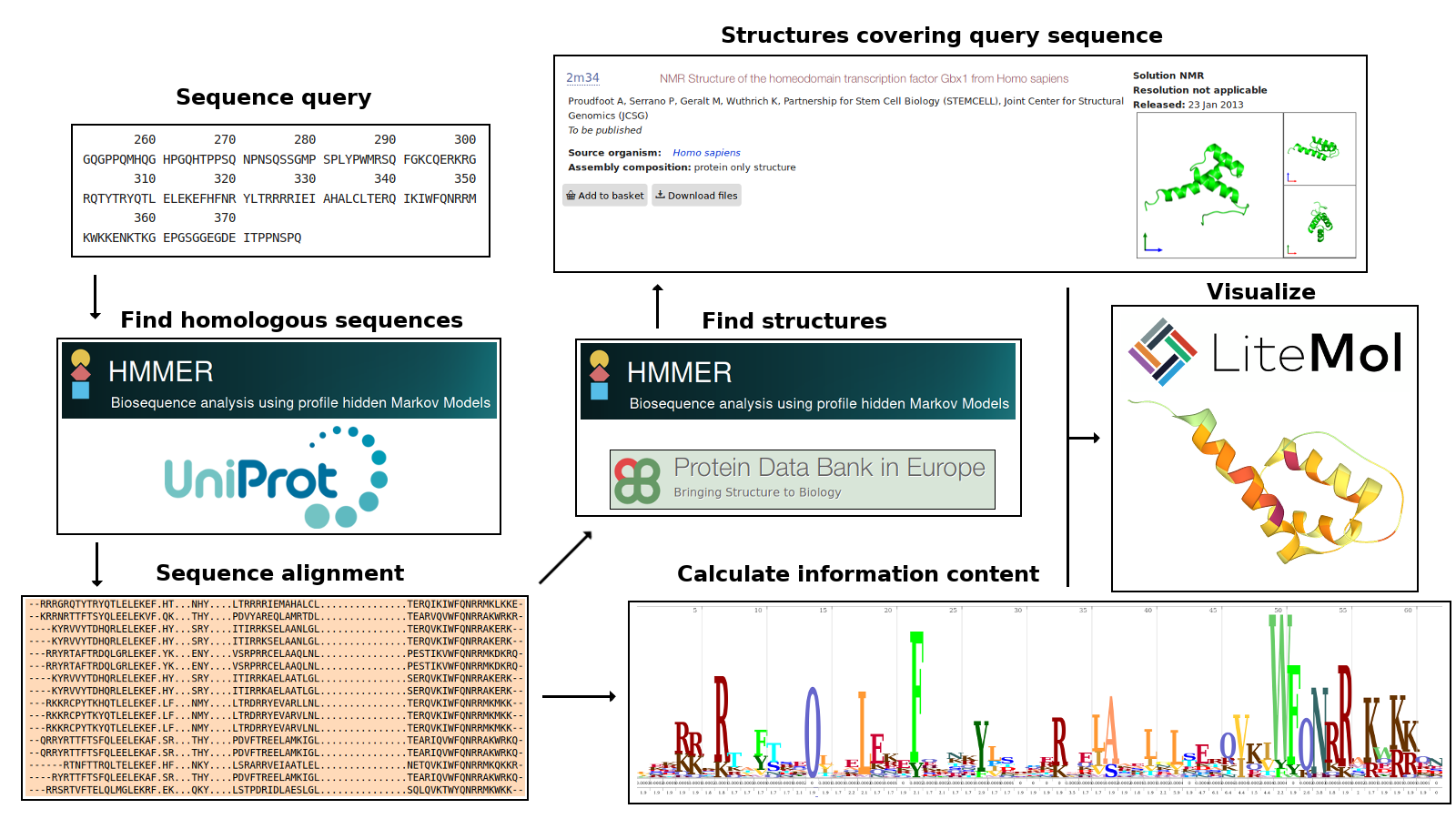
Input
Input to 3DPatch can be either a single protein sequence or a profile HMM file. These inputs must be in formats recognized by the immediately called HMMER programs (phmmer or hmmsearch, respectively). Refer to the HMMER web server documentation for detailed information on format requirements.
When sequence input is used, all whitespace characters are removed from the sequence before phmmer is called. If a header line beginning with the > symbol is found preceding the sequence, then the line is ignored.
Search parameters
All search parameters currently take on default values selected by the HMMER programs, the HMMER web server, and the Skylign server. HMMER web server default settings take precedence over those used by HMMER programs.
In the case of the sequence input route, the initial phmmer search against the UniProt reference proteomes uses a profile HMM with position-independent parameters derived from the BLOSUM62 substitution scoring matrix. In this profile HMM, the position-independent gap opening and gap extension probabilities are set to 0.02 and 0.4, respectively.
Per-target and per-domain reporting E-value thresholds are set to 1.0 for both phmmer and hmmsearch. Per-target and per-domain inclusion (significant hit) E-value thresholds are set to 0.01 and 0.03, respectively.
No taxonomy-based filtering of hits is being performed at any stage.
Saving and loading state
Save point files can be created once necessary information is available. All save point files contain the profile HMM information content profile, the list of PDB structures illustrating coverage of the profile HMM, and several variables necessary for state recreation. Optionally, if a structure was visualized at the time of save point file creation, it will be immediately shown in the LiteMol plugin after loading. Note that it is also possible to save the view of a structure not included in the profile HMM structure coverage illustration this way.
Loading state from a save point file recreates the profile HMM information content profile graph, the profile HMM structure coverage illustration, and the sequence alignment window. Optionally, the previously selected 3D structure is visualized.
It is possible to select a new structure from the profile HMM structure coverage illustration for information content-based mark-up when loading state from a save point file.
Some actions are disabled when loading state from a save point file. New search using a sequence or a profile HMM file input must be performed to have them enabled. These actions are:
- Selection of 3D structures not included in the profile HMM structure coverage illustration, or, optionally, the structure selected from the drop-down list of other domain hits at the time of save point file creation.
- Changing the color scaling option.
- Creation of new save point files.
Sequence alignment
Contents of this window vary depending on the type of input. If sequence input was used, then the consensus sequence representing the profile HMM built from the phmmer search results, the input sequence aligned to this profile HMM, and optionally the alignment of the selected PDB chain sequence region will be shown. If a profile HMM file input was used, then there will be no input sequence alignment.
The profile HMM consensus sequence is shown on the line starting with CONS. It contains one residue symbol per profile HMM position. The consensus sequence is read either from the profile HMM built by the HMMER web server from the phmmer search results, or from the uploaded profile HMM file.
If sequence input route was utilized, then the alignment of the input sequence to the profile HMM built from the phmmer search results is shown on the line starting with INPUT.
If some PDB structure is selected from the profile HMM structure coverage window for information content-based mark-up, the sequence of the marked-up region aligned to the profile HMM, and optionally to the input sequence, is shown on the line starting with the PDB chain ID. User can hover the mouse pointer over individual residues in this sequence to see them highlighted in the LiteMol viewer 3D structure visualization. When the selected residue is present in the 3D structure, its background color changes to magenta; if it is missing from the 3D structure, the background color changes to blue.
Note that residues assigned to insert states are not shown in the sequence alignment window.
Profile HMM structure coverage
When hmmsearch is called and the PDB is used as a sequence database, a list of PDB chains containing a hit to the profile HMM is returned. Each PDB chain can contain multiple sequence regions (called domains in HMMER terminology and in this section) aligned to the profile HMM. A score is calculated for each of these domain alignments. E-value reflecting the scores of individual domain alignments is assigned to each PDB chain. The list of PDB chains returned by hmmsearch is sorted by ascending chain E-value.
3DPatch goes through the list of PDB chain hits, starting with the chain with the lowest assigned E-value. For each chain, domains recognized within are iterated through in the order of their proximity to the protein N-terminus, starting with the most N-terminal one.
For each domain, a list profile HMM positions involved in the alignment of the domain sequence to the profile HMM is generated. If the profile HMM structure coverage illustration currently contains no domains, or if the number of new (previously uncovered) profile HMM positions covered by the alignment of the current domain is at least N, then the domain is added to the profile HMM structure coverage illustration, and the list of profile HMM positions covered by 3D structures is updated. The value of N is currently set to 40 positions.
If a domain does not meet the condition stated above, it does not appear in the profile HMM structure coverage illustration. However, it can still be visualized by manually selecting it from the drop-down list of all other domain hits. Each domain in the list is characterized by the PDB chain ID, the span of chain sequence residues involved in the alignment, and the span of profile HMM positions covered by the alignment. In addition, significance of the domain hit is graphically illustrated based on its independent E-value: 🔥 for E-values below 1e-20, *** for E-values between 1e-20 (inclusive) and 1e-10, ** for E-values between 1e-10 (inclusive) and 1e-2, and * for E-values greater than or equal to 1e-2.
Note that PDB chains are not treated equally by the HMMER web server. When multiple chains have the same amino acid sequence, they would yield the same domain hits spanning the same sets of residues. Each target considered by hmmsearch corresponds to a unique amino acid sequence, which can be shared by one or more PDB chains. The HMMER web server uses PDB chain ID of one of these chains as the accession attribute of the target sequence. 3DPatch makes it possible to also visualize domains found in chains other than the target's accession. These domains are added to the drop-down menu immediately after the corresponding domain recognized within the accession PDB chain had been added to the profile HMM structure coverage illustration or to the drop-down list. A table of accession PDB chains along with a comma-separated list of other PDB chains sharing the same amino acid sequence can be accessed from the 3DPatch website after the searches had finished. It may not be possible to display this table in some browsers when an advertisement blocking software is installed. It is not possible to select 3D structures from the drop-down menu when state is loaded from a save point file, with the exception of the structure selected at the time of save point file creation.
Color scaling options
3DPatch currently implements four information content color scaling options. Each of these options partitions the information content range into ten bins and assigns a color from the color spectrum to all information content values falling to a specific bin. The options differ by the transformation applied to individual information content values and by the value taken as the information content maximum. These options are:
- Linear (absolute), which uses maximum theoretical information content (appr. 6.45) as the maximum. Information content range is split evenly into ten bins, each covering around 0.65 bits.
- Linear (relative), which uses maximum observed information content as the maximum. Information content range is split evenly into ten bins, each covering one tenth of the maximum observed information content value. Note that the maximum observed value is calculated across all positions in the profile HMM, not only those covered by the currently visualized PDB structure. This is the default option.
- Exponential (absolute), which uses exp(maximum theoretical information content), appr. 634.68, as the maximum. The range is split evenly into ten bins, each covering around 63.47 units. Values of information content for which exp(information content) falls into the the range of the bin are assigned the corresponding color. For example, the first color is assigned to positions with information content between log(0) and log(63.47), i.e., between 0 (-inf) and 4.15 bits.
- Exponential (relative), which is the same as Exponential (absolute), but uses exp(maximum observed information content) as the maximum.
The color scaling option used can be changed after the calculations had finished without need to repeat preceding HMMER searches. Note that changing the color scaling option is not allowed when loading state from a save point file.
Coloring of residues outside the alignment region and residues assigned to insert states (green) and residues belonging to other chains (light blue) is unaffected by the color scaling choice.
Misc
It can happen that the 3DPatch process hangs when the HMMER servers are overloaded. You may want to reload the page and repeat the search, or try again later.
The PDB sequence database used by the HMMER web server may not be fully synchronized with the PDBe. Newly added structures may not appear in the list of hits, and structures made obsolete recently may fail to visualize.